Rare Metabolic Bone Disorders:
Overview:
Metabolic bone disorders are most commonly caused as a result of genetic abnormalities that directly or indirectly affect the bone or can alter bone cell function. These disorders often result in abnormal development and growth of the skeleton and may result in inability to maintain healthy bones. These disorders are rare and are inheritable, therefore, can be passed on to the children of affected adults. These disorders may also develop after birth from other medical problems and hence may not always be inherited. These disorders may also be present in children with unaffected adults; hence it becomes important to study these disorders in detail and understand the physiology, and inheritance of these diseases.
A few examples of Rare Metabolic Bone disorders:

Osteogenesis Imperfecta: Osteogenesis imperfecta (OI), also known as brittle bone disease, is a rare genetic disorder affecting approximately 1 in 10,000 individuals. The most common types of OI are caused by mutations in the type I collagen genes COL1A1 or COL1A2 and are inherited in an autosomal dominant manner. Other, rarer, types of OI are caused by mutations in genes involved in cross-linking, hydroxylation, or mineralisation of type I collagen and are inherited in an autosomal recessive manner. Some cases of OI are not inherited at all but are caused by de novo mutations.
Hypophosphatasia: Hypophosphatasia (HPP) is primarily thought of as a childhood disorder with soft bones caused by poor skeletal mineralization with a wide range of severity. Typically, the earlier the onset, the more severe the disease. Signs and symptoms may improve during the late teens through early adulthood only to recur in the 4th and 5th decades of life. In adults, HPP is typically associated with low bone mass, fractures, and tooth loss throughout adulthood. Joint pain and dysfunction are common and calcium can accumulate in soft tissues, hardening the tendons and ligaments, interfering with movement. Fractures are typically seen with soft bones, often in the feet, from poor skeletal mineralization (osteomalacia). Hypophosphatasia is caused by mutations in the ALPL gene, which encodes a protein, alkaline phosphatase, necessary for bone mineralization. The complete absence of alkaline phosphatase leads to the accumulation of a molecule that blocks bone mineralization and is typically lethal or severe early in life.
Tumor-induced osteomalacia: Tumor-induced osteomalacia (TIO; osteomalacia = soft bones), also known as oncogenic osteomalacia, is a rare form of low blood phosphorus (hypophosphatemia) that can present anytime in life. Individuals with TIO have bone pain, stress fractures, and muscle weakness. Stress fractures are caused by poor bone mineralization (osteomalacia), from low blood phosphorus. Multiple fractures can cause other deformities and even bowing of the legs. TIO is caused by a small tumor that produces extra amounts of FGF23, a hormone that decreases phosphorus levels in the blood. TIO is diagnosed based on the clinical findings and biochemical tests (low phosphorus, high FGF23, and other tests).
Fibrous dysplasia: Fibrous dysplasia (FD) is a disorder in which the bone is replaced by abnormal scar-like (fibrous) connective tissue. This abnormal fibrous tissue weakens the bone, making it abnormally fragile and prone to fracture. This disease is commonly observed in children or young adults and may remain undiagnosed in mild cases. As children grow, their bones become dysplastic. This disorder may manifest as monostotic (affecting one solitary bone) or polyostotic (affecting multiple bones throughout the body).
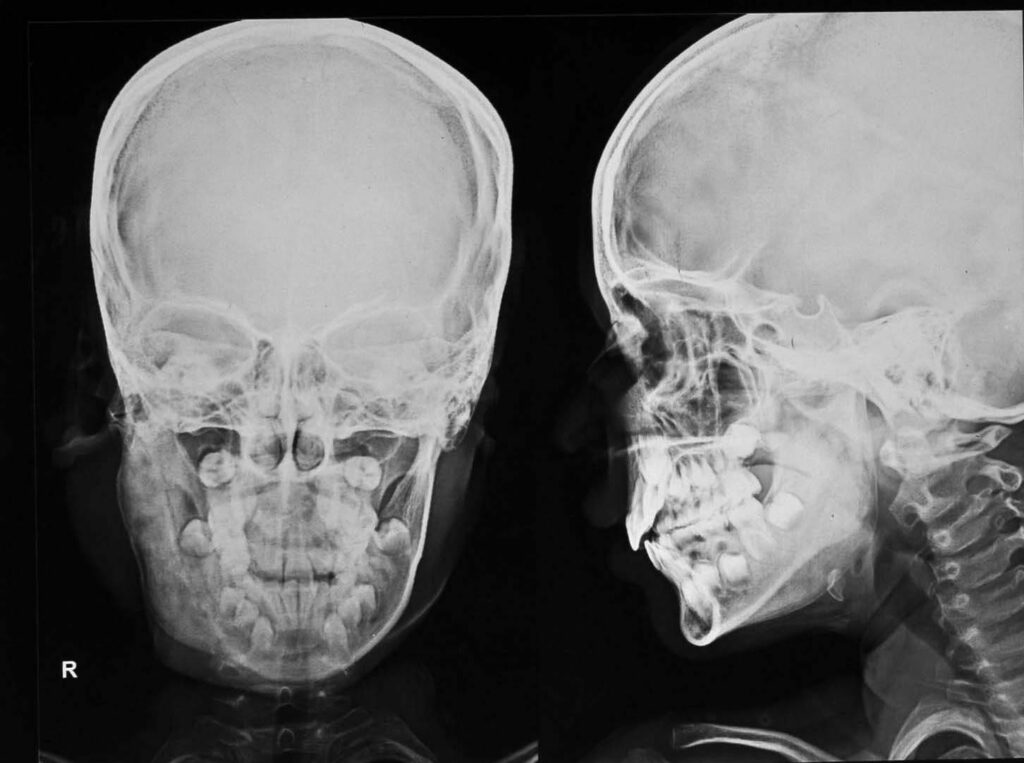
The severity of this condition generally varies from one person to the other. Most often, long bones, bones of the face and skull and ribs are affected. FD is a benign (noncancerous) disorder and does not spread. The bone or bones that are affected by the disorder are usually established early in life and it is very rare for new areas to become affected. The areas affected may be described as lesions. FD lesions may progressively grow and expand until an affected bone finishes growing. These lesions can eventually cause affected bones to become abnormally weakened, misshapen, and prone to facture. Bone pain can also occur and may be severe in some patients. The underlying cause of FD is not fully understood. Researchers believe that the disorder is caused by a change (mutation) in a gene called GNAS1. This gene mutation occurs after fertilization of the embryo (somatic mutation) and is therefore not inherited, nor will affected individuals pass the mutation on to their children.
Paget’s disease: Paget’s disease of bone is a chronic disease of the skeleton. In healthy bone, a process called remodelling removes old pieces of bone and replaces them with new, fresh bone. Paget’s disease causes this process to shift out of balance, resulting in new bone that is abnormally shaped, weak, and brittle. Paget’s disease most often affects older people, occurring in approximately 2 to 3 percent of the population over the age of 55. Many patients with Paget’s disease have no symptoms at all and remain undiagnosed. It most commonly manifests in the spine, pelvis, long bones of the limbs, and skull. It can be present in just one bone or in several bones in the body. The cause of Paget’s disease of bone is unknown. Research suggests a combination of environmental and genetic factors contribute to the disease. Several genes appear to be linked to getting the disease. Paget’s disease can lead to several complications such as: fractures and deformities, osteoarthritis, neurological problems and bone cancer.

Osteopetrosis: Osteopetrosis is a bone disease that makes bones abnormally dense and prone to fractures. Osteopetrosis is classified based on its inheritance pattern Autosomal dominant osteopetrosis (ADO), which is also called Albers-Schönberg disease, is typically the mildest type of the disorder. Some affected individuals have no symptoms. In these people, the unusually dense bones may be discovered by accident when an x-ray is done for another reason.

In affected individuals who develop signs and symptoms, the major features of the condition include multiple bone fractures, abnormal side-to-side curvature of the spine. Autosomal recessive osteopetrosis (ARO) is a more severe form of the disorder that becomes apparent in early infancy. Affected individuals have a high risk of bone fracture resulting from seemingly minor bumps and falls. Their abnormally dense skull bones pinch nerves in the head and face often resulting in vision loss, hearing loss, and paralysis of facial muscles. Dense bones can also impair the function of bone marrow, preventing it from producing new blood cells and immune system cells. As a result, people with severe osteopetrosis are at risk of abnormal bleeding, a shortage of red blood cells and recurrent infections. Mutations in the CLCN7 gene are responsible for about 75 percent of cases of autosomal dominant osteopetrosis ,and TCIRG1 gene mutations cause about 50 percent of cases of autosomal recessive osteopetrosis. Mutations in other genes are less common causes of autosomal dominant and autosomal recessive forms of the disorder.
Osteoporosis: Osteoporosis results in increased porosity of bones and causes bones to become weak and brittle. This increase in porosity makes patients diagnosed with osteoporosis highly prone to fragility fractures-which may occur due to falls or very mild stress. Osteoporosis affects both men and women, however, post-menopausal women are at a very high risk of sustaining fragility fractures due to osteoporosis. Osteoporosis-related fractures most commonly occur in the hip, wrist or spine. Age, sex, comorbidities, concomitant medications, race and lifestyle choices are common risk factors for osteoporosis. Bone fractures, particularly in the spine or hip, are the most serious complications of osteoporosis. Hip fractures often are caused by a fall and can result in disability and even an increased risk of death within the first year after the injury.
